хімічныя злучэнні, у састаў якіх уваходзіць медзь. Найб. пашыраныя злучэнні адна- і двухвалентнай медзі: медзі аксіды і солі мінер., а таксама некаторых карбонавых кіслот. Солі аднавалентнай медзі бясколерныя рэчывы, не раствараюцца ў вадзе, лёгка акісляюцца; двухвалентнай — раствараюцца ў вадзе, растворы маюць блакітны колер, які абумоўлены ўтварэннем аквакатыёнаў [Cu(H2O)4]2+. Солі ўтвараюць устойлівыя комплексныя злучэнні з шэрагам малекул і іонаў (напр., аміякаты).
Медзі (II) ацэтату монагідрат, ці мядзянка (CH3COO)2Cu∙H2O — цёмна-зялёныя крышталі. Выкарыстоўваюць як фунгіцыд, пігмент для керамікі, каталізатар полімерызацыі (напр., стыролу), стабілізатар штучных валокнаў. Медзі (II) карбанаты ўтвараюцца пры абменных рэакцыях у водных растворах паміж солямі Cu (II) і карбанатамі інш. металаў; з раствору ў залежнасці ад т-ры і канцэнтрацыі рэагентаў вылучаюцца нерастваральныя асноўныя, ці гідроксакарбанаты: дыгідроксакарбанат CuCO3∙Cu(OH)2 або Cu2(OH)2CO3 (у прыродзе мінерал малахіт) і дыгідроксадыкарбанат 2CuCO3∙Cu(OH)2 або Cu3(OH)2(CO3), (мінерал азурыт). Сярэдні карбанат медзі CuCO3 атрымліваюць апрацоўкай асн. карбанатаў дыаксідам вугляроду пад ціскам пры т-ры 180 °C. Медзі (II) сульфат CuSO4 — бясколерныя крышталі, tпл 200 °C, пры награванні (каля 650 °C) раскладаюцца. Утварае шэраг гідратаў, найважнейшы — пентагідрат, ці медны купарвас CuSO4∙5H2O — сінія крышталі, абязводжваюцца пры т-ры 250 °C. У прыродзе — мінерал халькантыт. У прам-сці атрымліваюць узаемадзеяннем медзі ці яе аксіду CuO з сернай к-той, абпалам сульфідаў медзі ў прысутнасці кіслароду. Выкарыстоўваюць як пігмент у фарбах, у сельскай гаспадарцы для барацьбы са шкоднікамі і хваробамі раслін і для пратручвання зерня (гл.Медныя ўгнаенні), для вырабу скуры, у гальванатэхніцы і інш.Медзі сульфіды: сульфід медзі (I), ці гемісульфід Cu2S і сульфід медзі (II), ці монасульфід CuS. Чорныя крышталі, не раствараюцца ў вадзе, раствараюцца ў азотнай кіслаце. У прыродзе трапляюцца ў выглядзе мінералаў хальказіну (Cu2S) і кавеліну (CuS). Усе М.з. атрутныя: раздражняюць слізістыя абалонкі, пашкоджваюць страўнікава-кішачны тракт, выклікаюць млоснасць, ірвоту, захворванне печані і інш.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
МЫШ’ЯКУ́ ЗЛУЧЭ́ННІ,
хімічныя злучэнні, у састаў якіх уваходзіць мыш’як. Найб. шырока выкарыстоўваюць аксіды і халькагеніды мыш’яку, арсеніды і шматлікія мыш’якарганічныя злучэнні.
Мыш’яку аксіды — злучэнні мыш’яку з кіслародам. Сэсквіаксід (мыш’яковісты ангідрыд ці белы мыш’як) As2O3 — белае цвёрдае рэчыва. Пры растварэнні ў вадзе ўтварае не вылучаныя ў свабодным стане ортамыш’яковістую H3AsO3 і металамыш’яковістую HAsO2 к-ты; пры ўзаемадзеянні са шчолачамі ўтварае арсеніты. Тэхн. атрымліваюць акісляльным абпалам сульфідных мінералаў мыш’яку. Выкарыстоўваюць для атрымання мыш’яку і яго злучэнняў, кансервавання скуры і футра, у вытв-сці аптычнага шкла, як інсектыцыд і некратызавальны лек. сродак. Аксід мыш’яку(V), ці мыш’яковы ангідрыд As2O5 — бясколерныя крышталі. Пры награванні раскладаецца на As2O3 і кісларод. Добра раствараецца ў вадзе, утварае ортамыш’яковую к-ту H3AsO4, солі якой наз.арсенатамі. Выкарыстоўваюць як гербіцыд, антысептык для прамочвання драўніны. Мыш’яку гідрыд (арсін, мыш’яковісты вадарод) AsH3 — газ без колеру і паху (часам мае часночны пах, абумоўлены наяўнасцю прадуктаў частковага акіслення AsH3). Пры т-ры каля 500 °C раскладаецца. Выкарыстоўваюць для атрымання мыш’яку высокай чысціні, легіравання паўправадніковых матэрыялаў мыш’яком. Мыш’яку халькагеніды, злучэнні мыш’яку з серай — сульфіды As2S3 (у прыродзе — мінерал аўрыпігмент), As4S4 (мінерал рэальгар), As4S3 (мінерал дымарфіт) i As2S5, з селенам — селеніды As2Se3 і As4Se4, з тэлурам — тэлурыд As2Te3. Усе халькагеніды, акрамя As2S5 (аморфнае рэчыва аранжавага колеру, крышталізуецца пад высокім ціскам), крышт. рэчывы. Устойлівыя ў паветры, не раствараюцца ў вадзе, добра раствараюцца ў растворах шчолачаў. As2S3, As2Se3 i As2Te3 — паўправаднікі. Атрымліваюць сплаўленнем элементаў у вакууме ці інертным асяроддзі. Выкарыстоўваюць як кампаненты халькагеніднага шкла, для вырабу валаконных святлаводаў у інфрачырв. вобласці спектра і інш. Усе растваральныя ў вадзе і слабакіслым асяроддзі (напр., страўнікавы сок) М.з. надзвычай атрутныя; злучэнні As(III) больш атрутныя за злучэнні As(V), асабліва небяспечныя AsH3 і AS2O3. ГДК мыш’яку і М.з. у паветры (у пераліку на мыш’як) 0,5 мг/м³, для AsH3 — 0,1 мг/м³.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
БЯЛКІ́, пратэіны,
прыродныя высокамалекулярныя арган. рэчывы, малекулы якіх складаюцца з астаткаў амінакіслот. Адзін з асн.хім. кампанентаў абмену рэчываў і энергіі жывых арганізмаў. Абумоўліваюць іх будову, гал. адзнакі, функцыі, разнастайнасць і адаптацыйныя магчымасці, удзельнічаюць ва ўтварэнні клетак, тканак і органаў (структурныя бялкі), у рэгуляцыі абмену рэчываў (гармоны), з’яўляюцца запасным пажыўным рэчывам (запасныя бялкі). Складаюць матэрыяльную аснову амаль усіх жыццёвых працэсаў: росту, стрававання, размнажэння, ахоўных функцый арганізма (гл.Антыцелы, Імунаглабуліны, Таксіны), утварэння генет. апарату і перадачы спадчынных прыкмет (нуклеапратэіды), пераносу ў арганізме рэчываў (транспартныя бялкі), скарачэнняў мышцаў, перадачы нерв. імпульсаў і інш.; ферменты бялковай прыроды выконваюць у арганізме спецыфічныя каталітычныя функцыі, выключна важнае значэнне ў рэгуляцыі фізіял. працэсаў маюць бялкі.-гармоны. Сінтэзуюцца бялкі з неарган. рэчываў раслінамі і некат. бактэрыямі. Жывёлы і чалавек атрымліваюць гатовыя бялкі з ежы. З прадуктаў іх расшчаплення (пептыдаў і амінакіслот) у арганізме сінтэзуюцца спецыфічныя ўласныя бялкі, дзе яны няспынна разбураюцца і замяняюцца зноў сінтэзаванымі. Біясінтэз бялкоў ажыццяўляецца па матрычным прынцыпе з удзелам ДНК, РНК, пераважна ў рыбасомах клетак і інш. Паслядоўнасць амінакіслот у бялках адлюстроўвае паслядоўнасць нуклеатыдаў у нуклеінавых к-тах. Паводле паходжання і крыніц атрымання бялкоў падзяляюцца на раслінныя, жывёльныя і бактэрыяльныя, паводле хім. саставу — на простыя (некан’югіраваныя) — пратэіны і складаныя (кан’югіраваныя) — пратэіды. Простыя складаюцца з астаткаў амінакіслот, што злучаны паміж сабою пептыднай сувяззю (—NH—CO) у доўгія ланцугі — поліпептыды, складаныя — з простага бялку, злучанага з небялковым арган. ці неарган. кампанентам непептыднай прыроды, т.зв.прастэтычнай групай, далучанай да поліпептыднай часткі. Сярод складаных бялкоў паводле тыпу прастэтычнай групы вылучаюць нуклеапратэіды, фосфапратэіды, глікапратэіды, металапратэіды, гемапратэіды, флавапратэіды, ліпапратэіды і інш. У састаў бялкоў уваходзіць ад 50 да 6000 і больш астаткаў 20 амінакіслот, што ўтвараюць складаныя поліпептыдныя ланцугі. Амінакіслотны састаў розных бялкоў неаднолькавы і з’яўляецца іх важнейшай характарыстыкай, а таксама мерай харч. каштоўнасці. Паслядоўнасць амінакіслот у кожным бялку вызначаецца паслядоўнасцю монануклеатыдных буд. блокаў у асобных адрэзках малекулы ДНК. Вядома амінакіслотная паслядоўнасць некалькіх соцень бялкоў (напр., адрэнакортыкатропнага гармону чалавека, рыбануклеазы, цытахромаў, гемаглабіну і інш.). Парушэнні амінакіслотнай паслядоўнасці ў малекуле бялку выклікаюць т.зв.малекулярныя хваробы. Амінакіслотную паслядоўнасць поліпептыднага ланцуга для малекулы гармону інсуліну ўстанавіў англ. біяхімік Ф.Сэнгер (1953). Звесткі пра колькасць адрозненняў у амінакіслотных паслядоўнасцях гамалагічных бялкоў, узятых з розных відаў арганізмаў, выкарыстоўваюць пры складанні эвалюцыйных картаў, якія адлюстроўваюць паслядоўныя этапы ўзнікнення і развіцця пэўных відаў арганізмаў у працэсе эвалюцыі.
Агульны хім.састаў бялкоў (у % у пераліку на сухое рэчыва): C — 50—55, O — 21—23, N — 15—18, H — 6—7,5, S — 0,3—2,5, P — 1—2, і інш. Малекулярная маса ад 5 тыс. да 10 млн. Большасць бялкоў раствараецца ў вадзе і ўтварае малекулярныя растворы. Па форме малекул адрозніваюць бялкі фібрылярныя (ніткападобныя) і глабулярныя (згорнутыя ў кампактную структуру сферычнай формы); па растваральнасці ў вадзе, растворах нейтральных соляў, шчолачах, кіслотах і арган. растваральніках вылучаюць альбуміны, гістоны, глабуліны, глютэліны, праламіны, пратаміны і пратэіноіды. Бялкі маюць кіслыя карбаксільныя і амінныя групы, таму ў растворах яны амфатэрныя (маюць уласцівасці асноў і к-т). Пры гідролізе яны распадаюцца да амінакіслот; пад уплывам розных фактараў здольныя да дэнатурацыі і каагуляцыі, уступаюць у рэакцыі акіслення, аднаўлення, нітравання і інш. Пры пэўных значэннях pH у растворах бялкоў пераважае дысацыяцыя тых ці інш. груп, што надае ім адпаведны зарад і выклікае рух у электрычным полі — электрафарэз. Структура бялкоў характарызуецца амінакіслотным саставам, парадкам чаргавання амінакіслотных астаткаў у поліпептыдных ланцугах, іх даўжынёй і размеркаваннем у прасторы. Адрозніваюць 4 парадкі (узроўні) структуры бялкоў: першасную (лінейная паслядоўнасць амінакіслотных астаткаў у поліпептыдным ланцугу), другасную (прасторавая, найчасцей спіральная прасторавая канфігурацыя, якую прымае сам поліпептыдны ланцуг), трацічную (трохмерная канфігурацыя, якія ўзнікае ў выніку складвання або закручвання структур другаснага парадку ў больш кампактную глабулярную форму) і чацвярцічную (злучэнне некалькіх частак з трацічнай структурай у адну больш буйную комплексную праз некавалентныя сувязі). Найб. устойлівая першасная структура бялкоў, іншыя лёгка разбураюцца пры павышэнні т-ры, рэзкім змяненні pH асяроддзя і інш. уздзеяннях (дэнатурацыя бялкоў), што вядзе да страты асн.біял. уласцівасцяў. Фарміраванне прасторавай канфігурацыі малекул бялку вызначаецца наяўнасцю ў поліпептыдных ланцугах вадародных, дысульфідных, эфірных і салявых сувязяў, сіл Ван дэр Ваальса і інш. Уласцівасці бялкоў залежаць ад іх хім. будовы і прасторавай арганізацыі (канфармацыі). Наяўнасць некалькіх узроўняў арганізацыі Б. забяспечвае іх вял. разнастайнасць у прыродзе (напр., у клетках бактэрыі Escherichia coli каля 3000 розных бялкоў, у арганізме чалавека больш за 50 000). Кожны від арганізмаў мае ўласцівы толькі яму набор бялкоў, па якім ён можа быць індэнтыфікаваны. Органы і тканкі жывых арганізмаў маюць розную колькасць бялкоў (у % да сырой вагі); 6,5—8,5 у крыві, 7—9 у мозгу, 16—18 у сэрцы, 18—23 у мышцах, 10—20 у насенні злакаў, 20—40 у насенні бабовых, 1—3 у лісці большасці раслін. Па харч. каштоўнасці бялкі падзяляюць на паўнацэнныя (маюць усе амінакіслоты, неабходныя жывёльнаму арганізму для сінтэзу бялкоў сваіх тканак) і непаўнацэнныя (у складзе малекул няма некаторых амінакіслот). Сутачная патрэба дарослага чалавека ў бялках 100—120 г. Арганізм расходуе ўласныя бялкі, калі ў ежы іх менш за норму. Многія прыродныя бялкі і бялковыя ўтварэнні выкарыстоўваюць у прам-сці (напр., для вырабу скуры, шэрсці, натуральнага шоўку, казеіну, пластмасаў і інш.), медыцыне і ветэрынарыі (як лек. сродкі і біястымулятары, напр., інсулін пры цукр. дыябеце, сываратачны альбумін як заменнік крыві, гама-глабулін для прафілактыкі інфекц. захворванняў, бялкі-ферменты для лячэння парушэнняў абмену рэчываў, гідралізатары бялкоў для штучнага жыўлення). Для атрымання пажыўных і кармавых бялкоў выкарыстоўваюць мікрабіял. сінтэз. Вядуцца даследаванні па штучным сінтэзе бялковых малекул (штучна сінтэзаваны фермент рыбануклеаза і інш.). Бялкі — адзін з гал. аб’ектаў даследаванняў біяхіміі, імуналогіі і інш. раздзелаў біял. навукі.
Літ.:
Бохински Р. Современные воззрения в биохимии: Пер. с англ.М., 1987;
Ленинджер А. Основы биохимии: Пер. с англ.Т. 1—3. М., 1985;
Гершкович А.А. От структуры к синтезу белка. Киев, 1989;
Овчинников Ю.А. Химия жизни: Избр. тр.М., 1990.
У.М.Рашэтнікаў.
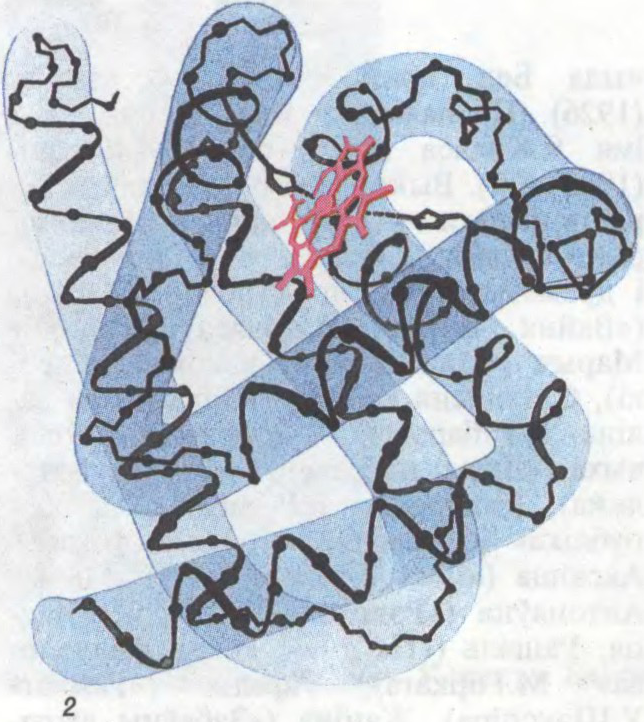
Да арт.Бялкі. Малекула бялку міяглабіну: 1 — агульны выгляд; 2 — структурная схема.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
БА́РЫЮ ЗЛУЧЭ́ННІ,
хімічныя злучэнні, у састаў якіх уваходзіць барый, пераважна ў ступені акіслення +2. Найб. пашыраны аксід, гідраксід барыю, солі барыю (сульфат, хларыд, карбанат, нітрат і інш.). Бясколерныя крышт. рэчывы, ядавітыя, ГДК амаль усіх барыю злучэнняў 0,5 мг/м³. Сыравінай у вытв-сці барыю злучэнняў з’яўляецца барытавы канцэнтрат (80—95% сульфату барыю), які атрымліваюць флатацыяй барыту.
Барыю аксід BaO, tпл 2017 °C, пры награванні ўзганяецца, шчыльн. 5,7∙103кг/м³. З вадой утварае гідраксід барыю, з кіслотамі, дыяксідам вугляроду — солі. Выкарыстоўваюць у вытв-сці шкла, эмаляў, каталізатараў. Пры награванні ў кіслародзе (500 °C) пераходзіць у пераксід барыю BaO2 — кампанент піратэхн. сумесяў, адбельвальнікаў для тканін і паперы. Барыю гідраксід Ba(OH)2, tпл 408 °C, гіграскапічны, насычаны раствор у вадзе наз. барытавай вадой; моцная аснова. Выкарыстоўваецца як паглынальнік дыяксіду вугляроду, для ачысткі алеяў і тлушчаў, кампанент змазак, аналітычны рэагент на сульфат- і карбанат-іоны. Барыю сульфат BaSO4, tпл 1580 °C, шчыльн. 4,5∙103кг/м³, не раствараецца ў вадзе і разбаўленых кіслотах, паглынае рэнтгенаўскае выпрамяненне. Напаўняльнік гумы і паперы (у т. л. фотапаперы), кардону, кампанент белых мінер. фарбаў, кантрастнае рэчыва ў рэнтгенаскапічных даследаваннях страўнікава-кішачнага тракту (ГДК 6 мг/м³). Барыю карбанат BaCO3, tпл 1555 °C (у атмасферы CO2 пад ціскам 45 МПа), шчыльн. 4,25∙103кг/м³. Дрэнна раствараецца ў вадзе, рэагуе з разбаўленымі салянай і азотнай кіслотамі. Трапляецца ў прыродзе як мінерал вітэрыт. Выкарыстоўваюць у вытв-сці катодаў у электронна-вакуумных прыстасаваннях, аптычнага шкла, эмаляў, палівы, керамічных матэрыялаў, ферытаў, чырв. цэглы. Барыю хларыд BaCl2, tпл 961 °C, шчыльн. 3,83∙103кг/м³, раствараецца ў вадзе. Выкарыстоўваецца ў гарбарнай прам-сці для ўцяжарвання і асвятлення скуры, для барацьбы са шкоднікамі ў сельскай гаспадарцы, загартоўкі «хуткарэзнай» сталі. Барыю нітрат Ba(NO3)2. Існуе як мінерал нітрабарыт, выкарыстоўваецца ў эмалях і паліве, піратэхніцы. Барыю тытанат BaTiO3 — сегнетаэлектрык. Барыю храмат BaCrO4 і манганат BaMnO4 — адпаведна жоўты і зялёны пігменты.
Літ.:
Ахметов Т.Г. Химия и технология соединений бария. М., 1974.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
АЛЮМІ́НІЮ ЗЛУЧЭ́ННІ,
хімічныя злучэнні, у састаў якіх уваходзіць алюміній, пераважна ў ступені акіслення + 3. Бясколерныя, белыя ці шэрыя цвёрдыя рэчывы. Найб. пашыраны алюмінію злучэнні з кіслародам (крышт.алюмінію аксід і аморфны алюмагель, гідраксід алюмінію), солі алюмінію з моцнымі кіслотамі (нітрат, сульфат, галагеніды, фасфаты), комплексныя солі алюмінію (алюмініевы галын, алюмасілікаты), солі алюмініевых кіслот (алюмінаты), алюмінійарган. злучэнні (гл. ў арт.Металаарганічныя злучэнні), нітрыд і гідрыд алюмінію, алюмінію злучэнні з некаторымі больш электрададатнымі, чым алюміній, металамі, напр. арсенід алюмінію.
Алюмінію гідраксід (Al(OH)3] сустракаецца ў прыродзе ў выглядзе мінералаў — састаўная частка баксітаў, існуе ў трох крышт. і аморфнай мадыфікацыях; не раствараецца ў вадзе, спіртах; амфатэрны, з кіслотамі ўтварае солі, са шчолачамі алюмінаты. Атрымліваюць гідролізам алюмасілікатаў у шчолачным асяроддзі, аморфны — асаджэннем з раствораў соляў алюмінію аміякам. Выкарыстоўваюць для вытв-сці аксіду алюмінію і алюмагелю, як адсарбцыйны сродак у медыцыне. Алюмінію сульфат [Al2(SO4)3], т-ра раскладання больш за 770 °C, раствараецца ў вадзе. Атрымліваецца ўзаемадзеяннем кааліну ці баксіту з сернай кіслатой. Выкарыстоўваюць у вытв-сці алюмініевага галыну, для праклейвання паперы, асвятлення і пазбаўлення колеру вады, як пратраву пры фарбаванні тканін. Алюмінію фтарыд (AlF3), т-ра ўзгонкі 1279 °C, раствараецца ў вадзе, утварае крышталегідраты. Кампанент электраліту ў вытв-сці алюмінію, таксама флюсаў, эмаляў, керамікі. Алюмінію хларыд (AlCl3), дыміць на паветры, т-ра ўзгонкі 180 °C, tпл 192,5 °C, у вадзе гідралізуецца. Атрымліваецца хларыраваннем кааліну, баксіту ці гліназёму. Каталізатар крэкінгу нафты, у рэакцыях алкіліравання. Алюмінію нітрыд (AlN), т-ра раскладання ~2000 °C, дыэлектрык, устойлівы да дзеяння кіслот і шчолачаў пры t 20 °C. Атрымліваюць узаемадзеяннем азоту з алюмініем пры t 1000 °C ці аднаўленнем аксіду алюмінію. Выкарыстоўваецца як вогнетрывалы матэрыял для тыгляў, футровак электролізных ваннаў, для нанясення каразійна- і зносаўстойлівых пакрыццяў на сталь, графіт і інш.Алюмінію гідрыд (AlH3), т-ра раскладання 105 °C, існуе ў палімерным стане. Выкарыстоўваецца як кампанент цвёрдага ракетнага паліва, аднаўляльнік у арган. Сінтэзе. Алюмінію арсенід (AlAs), т-ра плаўлення 1740 °C, кампанент паўправадніковых цвёрдых раствораў для лазераў, фотадыёдаў, сонечных батарэй.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
ГІДРАГЕАЛО́ГІЯ (ад гідра... + геалогія),
навука аб падземных водах, іх саставе, уласцівасцях, фарміраванні, пашырэнні, руху, узаемадзеянні з горнымі пародамі і паверхневымі водамі. Распрацоўвае метады вызначэння гідрагеал. параметраў ваданосных гарызонтаў, пошуку і разведкі радовішчаў падземных вод, ажыццяўляе ацэнку запасаў і рэсурсаў падземных вод рэгіёнаў, краіны, вывучае рэжым і баланс падземных вод, даследуе гідраўлічную сувязь паверхневых і падземных вод, водаабмен паміж ваданоснымі гарызонтамі і комплексамі, прагназуе змены гідралагічных умоў тэрыторыі пад уздзеяннем меліярацыі, водазабораў падземных вод і інш.
Першыя звесткі аб паходжанні і ўласцівасцях прыродных вод адносяцца да 1-га тыс. да н. э. (Стараж. Грэцыя — Фалес, Арыстоцель, Стараж. Рым — Лукрэцый). У эпоху Адраджэння і пазней у Зах. Еўропе падземныя воды вывучалі Г.Агрыкала, Б.Палісі, Н.Стэна і інш.; у Расіі — М.В.Ламаносаў, В.М.Севяргін. Да сярэдзіны 19 ст. вучэнне аб падземных водах было часткай геалогіі. Як самаст. навука гідрагеалогія сфарміравалася ў канцы 19 ст. У развіццё гідрагеалогіі значны ўклад зрабілі А.Дарсі, Ж.Дзюпюі, А.Шэзі (Францыя), Э.Прынц, К.Кайльгак, Г.Гёфер (Германія), А.Хазен, Ч.Сліхтэр, О.Мейнцэр (ЗША), А.П.Карпінскі, С.М.Нікіцін, І.В.Мушкетаў (Расія) і інш.
На Беларусі комплексныя гідрагеал. даследаванні арганізаваны ў 1928 (у бас.р. Свіслач), у выніку якіх выяўлены крыніцы водазабеспячэння г. Мінск. У 1929—35 дзейнічаў Ін-т геалогіі і гідрагеалогіі АНБССР, з 1935 сектар гідрагеалогіі і інж. геалогіі ў Ін-це геал. навук АН. У 1938 на базе свідравіны, якая ўскрыла мінер. воды ў Бабруйску, пабудавана першая на Беларусі водалячэбніца. Пасля Вял.Айч. вайны складзены кадастр падземных вод, дробнамаштабныя гідрагеал. карты Беларусі, вывучаны хімізм і асн. элементы балансу падземных вод. З 1958 вядзецца сярэднемаштабная геолага-гідрагеал., з 1970 гідрагеал. здымка. Рэжым, т-ру і хім.састаў падземных вод і расолаў, умовы іх фарміравання, прынцыпы аховы і рацыянальнага выкарыстання даследуюцца ў Ін-це геал. навук АН Беларусі (Г.В.Багамолаў, М.Ф.Казлоў, А.В.Кудзельскі), у Бел.н.-д. геолагаразведачным ін-це (А.П.Лаўроў, С.П.Гудак), Бел. геолага-гідрагеал. экспедыцыі, БДУ, Гомельскім дзярж. ун-це і інш.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
ЖАЛЕ́ЗА ЗЛУЧЭ́ННІ,
хімічныя злучэнні, у састаў якіх уваходзіць жалеза, пераважна ў ступені акіслення +2 і +3. Найб. пашыраны неарган. Ж.з. — жалеза аксіды, гідраксіды, карбіды (гл.Жалезавугляродзістыя сплавы), сульфіды, сульфаты, карбанілы (гл.Карбанілы металаў), цыяністыя комплексныя солі (гексацыянафераты, гл.Калію злучэнні), а таксама жалезаарган. злучэнні (гл.Ферацэн).
Жалеза (II) гідраксід Fe(OH)2 — бледна-зялёнае аморфнае ці крышт. рэчыва. Узаемадзейнічае з к-тамі, акісляецца на паветры да гідраксіду Fe (III). Жалеза (III) гідраксід Fe2O3nH2O — бурае аморфнае рэчыва, не раствараецца ў вадзе, раствараецца ў к-тах. У прыродзе — буры жалязняк. Асаджэннем шчолаччу з раствораў солей Fe (III) атрымліваюць Fe(OH)3 — слабая аснова, амфатэрны (пры сплаўленні са шчолачамі ўтварае ферыты). Выкарыстоўваюць як кампанент жоўтага пігменту для фарбаў і эмалей. Жалеза сульфаты, солі сернай к-ты — крышт., гіграскапічныя рэчывы, раствараюцца ў вадзе. Утвараюць крышталегідраты: гептагідрат FeSO4∙7H2O — жалезны купарвас (мінерал мелантэрыт), блакітнавата-зялёныя крышталі, tпл 64 °C. З сульфатамі шчолачных металаў і амонію ўтвараюць двайныя сульфаты: FeSO4∙(NH4)2SO4∙6H2O (соль Мора) — сіне-зялёныя крышталі, устойлівыя на паветры; NH4Fe(SO4)212H2O (жалеза-амоніевы галын). Выкарыстоўваюць як кампанент электраліту ў гальванатэхніцы; FeSO4 — кансервант драўніны, фунгіцыд, антыанемічны сродак і інш.; Fe2(SO4)3 — растваральнік у гідраметалургіі медзі, каагулянт пры ачыстцы вады, пратрава пры фарбаванні. Жалеза сульфіды — злучэнні жалеза з серай. Монасульфід жалеза FeS, рудыя ці чорныя крышталі, tпл 1193 °C. Не раствараецца ў вадзе, раскладаецца к-тамі. У прыродзе — мінералы пірацін (гл.Калчаданы) і траіліт. Выкарыстоўваюць для атрымання серавадароду. Дысульфід FeS2, залаціста-жоўтыя крышталі, мінералы пірыт і марказіт. Выкарыстоўваюць прыродны як сыравіну для атрымання серы, сернай к-ты, сульфату Fe2(SO4)3, сінт. — каталізатар у арган. сінтэзе. Жалеза хларыды — злучэнні жалеза з хлорам, солі салянай к-ты — крышт., гіграскапічныя рэчывы, раствараюцца ў вадзе, этаноле, ацэтоне. Дыхларыд жалеза FeCl2 утвараецца пры ўзаемадзеянні жалеза з салянай к-той. Выкарыстоўваюць для атрымання трыхларыду. Трыхларыд FeCl3, цёмна-рудыя крышталі. 309 °C. Выкарыстоўваюць як каагулянт пры ачыстцы вады, кампанент раствораў для электрахіміі, траўлення пячатных плат і інш.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
ЗАЦЬМЕ́ННІ,
астранамічныя з’явы, пры якіх нябесныя свяцілы часткова або поўнасцю робяцца нябачнымі. Адбываюцца з-за таго, што больш далёкае ад Зямлі нябеснае цела закрываецца больш блізкім, ці таму, што на адно нябеснае цела падае цень другога. Да З. адносяць сонечныя і месяцовыя З., а таксама закрыцці зорак і планет (Месяц пры руху закрывае зорку ці планету), праходжанні планет па дыску Сонца (назіраюцца ў Меркурыя і Венеры), З. спадарожнікаў іншых планет, праходжанні ценю спадарожніка па дыску планеты і інш. Звесткі аб момантах З. і ўмовах іх бачнасці прыводзяцца ў астр. штогодніках.
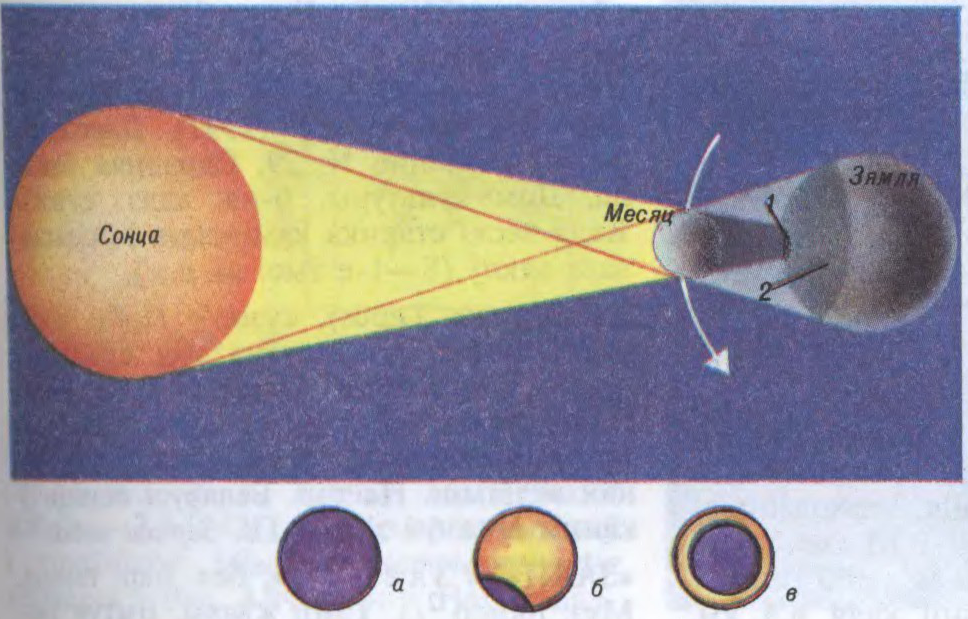
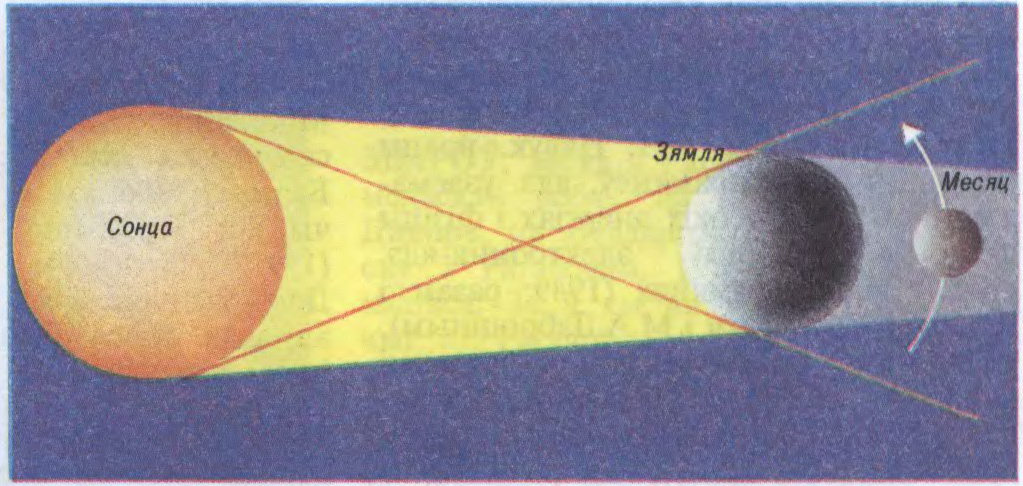
Сонечныя З. адбываюцца, калі Месяц (у фазе маладзіка), праходзячы паміж Зямлёю і Сонцам, поўнасцю ці часткова засланяе Сонца. Поўнае З. Сонца назіраецца там, дзе на Зямлю падае цень Месяца. Дыяметр ценю звычайна не перавышае 250—270 км. Месяц рухаецца, і яго цень перамяшчаецца і вычэрчвае паслядоўна вузкую паласу поўнага З. Фаза поўнага З. доўжыцца да 7 мін 30 с, найчасцей 2—3 мін. Па-за паласой, куды падае паўцень Месяца, назіраецца частковае З.
Сонца Калі бачны вуглавы дыяметр Месяца меншы за сонечны, назіральнік бачыць кольцападобнае З. У час сонечнага З. даследуюць дынаміку і спектральны састаў атмасферы Сонца, сонечную карону, праводзяць эксперыменты для праверкі эфектаў тэорыі адноснасці па адхіленні прамянёў святла, што ідуць ад далёкіх зорак паблізу Сонца ў полі яго прыцягнення. Месяцовыя З. адбываюцца, калі Месяц (у поўню) і Сонца знаходзяцца з процілеглых бакоў ад Зямлі і Месяц часткова ці поўнасцю трапляе ў цень Зямлі. Назіраюцца адначасова на ўсім паўшар’і Зямлі, павернутым да Месяца. Працягласць поўнага З. Месяца 1 гадз 4 мін, а ўсяго З. ад пачатку да канца — больш за 3 гадз. Месяц поўнасцю не знікае ў час З., а слаба бачны з прычыны сонечнага святла, што пераламляецца ў зямной атмасферы.
Літ.:
Дагаев М.М. Солнечные и лунные затмения. М., 1978.
Н.А.Ушакова.
Схема сонечнага зацьмення: 1 — зона поўнага зацьмення; 2 — зона частковага зацьмення; а, б, в — поўнае, частковае, кольцападобнае зацьменні.Схема зацьмення Месяца.Да арт.Зацьменні. Сонечная карона, сфатаграфаваная ў час сонечнага зацьмення.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
МА́НТЫЯ ЗЯМЛІ́,
сілікатная абалонка «цвёрдай» Зямлі паміж зямной карой і ядром Зямлі; адна з геасфер. Складае 83% аб’ёму і 67% масы Зямлі. Звесткі пра будову і стан рэчыва М.З. атрымліваюць шляхам сейсмалагічных назіранняў. Верхняя мяжа праходзіць на глыб. ад некалькіх кіламетраў (пад акіянамі) да 75 км (пад кантынентамі) па Махаровічыча паверхні і характарызуецца перападам скорасці сейсмічных хваль ад 6,4—7 км/с у зямной кары да 8—8,4 км/с у падкоравым слоі мантыі; ніжняя — на глыб. каля 2,9 тыс.км на мяжы з ядром Зямлі, з перападам скорасці хваль ад 13,25 км/с у мантыі да 8,5 км/с у верхняй частцы ядра. Мантыя падзяляецца на верхнюю (слой B) да глыб. 400 км, сярэднюю (слой C, слой Б.Б.Галіцына) да глыб. 950—1 тыс.км (паводле інш. меркаванняў да 900 км), дзе назіраецца рэзкае нарастанне хуткасцей сейсмічных хваль з глыбінёй, і ніжнюю (слой Д), дзе скорасці манатонна павялічваюцца да падэшвы. Т-ра М.З. 2000—2500 °C. Шчыльн. рэчыва М.З. з глыбінёй нарастае ад 3,3—3,4 г/см³ у падкоравым слоі да 3,65—3,7 г/см³ на глыб. 400 км, у слоі C — рэзка нарастае да 4,55—4,65 г/см³ на глыб. 950—1 тыс.км, потым павольна павялічваецца да 5,55—5,65 г/см³ каля падэшвы М.З. Ціск у нізе М.З. (1,35—1,40)∙1011 Па, мяркуецца, што пры высокіх цісках рэчыва мантыі знаходзіцца ў цвёрдым крышталічным стане, акрамя астэнасферы, дзе яно, магчыма, аморфнае. Сярэдняя вязкасць рэчыва М.З. каля 1023—1025 П, у астэнасферы пад акіянамі 1019—1020, пад кантынентамі 1021—1022 П. У версе М.З. складзена з лерцалітаў, перыдатытаў, эклагітаў, ніжэй — з піралітаў. Па супастаўленні геафіз. і мінер. даных мяркуецца, што паводле мінералаг. складу М.З. падзяляецца на верхнюю — алівінавую, сярэднюю — шпінеле-пераўскіта-ільменітавую. дзе вылучаюць 2 зоны на глыб.,420 км і 670 км, і ніжнюю — пераўскітавую. У М.З. існуюць канвектыўныя патокі рэчыва, аб чым, магчыма, сведчыць дрэйф літасферных пліт, якія неаднаразова перамешвалі састаў верхняй і ніжняй мантыі. З працэсамі ў М.З. звязаны тэктанічныя рухі, магматызм, вулканізм і інш.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
МІНЕРАЛО́ГІЯ (ад позналац. minera руда + ...логія),
навука аб прыродных хім. злучэннях — мінералах. Вывучае састаў, уласцівасці, марфалогію, структуру, працэсы ўтварэння і змянення мінералаў, заканамернасці сумеснага знаходжання ў прыродзе, а таксама ўмовы і метады штучнага атрымання з мэтай іх практычнага выкарыстання. Уваходзіць у комплекс геал. навук і цесна звязана з петраграфіяй, крышталяграфіяй, геахіміяй, вучэннем аб карысных выкапнях і інш. Аб’ект даследавання ў М. — асобныя крышталі, іх агрэгаты, генетычныя сукупнасці і інш. У 2-й пал. 20 ст. сфарміраваліся раздзелы М. касмічнай і М. мантыі. Вял. значэнне мае эксперыментальная М., якая займаецца мадэліраваннем фіз.-хім. працэсаў утварэння мінералаў, іх сінтэзам.
М. — найстаражытнейшая з навук геал. цыкла. Тэрмін «М.» ўведзены ў 1636 італьян. натуралістам Б.Цэзіем. М. развівалася паралельна з горнай справай і металургіяй. Элементы мінер. ведаў трапляюцца ў натурфілосафаў (з сярэдзіны 4 ст. да н.э.). Арыстоцель вылучаў у мінер. свеце 2 класы: камяні і руды. Класіфікацыяй мінералаў займаліся Тэафраст, Пліній Старэйшы, у 10—12 ст. Біруні, Ібн Сіна, Альберг Вялікі. Накапленне ведаў аб мінералах (у 17 ст. ў працах дацкіх вучоных Э.Барталіна, Н.Стэна, англ. Р.Бойля, Р.Гука, галандскага К.Гюйгенса, у 18—19 ст. — франц. Ж.Б.Рамэ дэ Ліля, Р.Ж.Гаюі, англ. У.Воластана, ням. А.Г.Вернера, рус. М.В.Ламаносава, В.М.Севергіна і інш.) прывяло да дыферэнцыяцыі М. і вылучэння з яе крышталяграфіі (18 ст.), петраграфіі (19 ст.), вучэння аб карысных выкапнях, геахіміі і металагеніі (канец 19 — пач. 20 ст.), вучэння аб каўстабіялітах (20 ст.), крышталяхіміі (сярэдзіна 20 ст.). Вял. ўклад у развіццё М. зрабілі рус. вучоныя М.І.Какшараў, П.У.Ерамееў, А.П.Карпінскі, Я.С.Фёдараў, сав. вучоныя А.Г.Бяцехцін, А.К.Болдыраў, У.І.Вярнадскі, А.М.Заварыцкі, У.М.Лодачнікаў, С.С.Смірноў, А.Я.Ферсман і інш.
На Беларусі мінералагічныя даследаванні праводзяцца паралельна з літалагічнымі, петраграфічнымі, з вывучэннем карысных выкапняў і стратыграфіі. Імі займаюцца ў ВА «Белгеалогія», Бел.н.-д. геолагаразведачным ін-це, Ін-це геал. навук Нац.АН Беларусі і інш.