БРОМ (
Br, хімічны элемент VII групы
Бром — чырвона-бурая вадкасць з непрыемным рэзкім пахам, лёгка выпараецца, tпл -7,25
Літ.:
Ксензенко В.И., Стасиневич Д.С.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
БРОМ (
Br, хімічны элемент VII групы
Бром — чырвона-бурая вадкасць з непрыемным рэзкім пахам, лёгка выпараецца, tпл -7,25
Літ.:
Ксензенко В.И., Стасиневич Д.С.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
КЛА́СТЭРЫ (
групоўкі хімічна злучаных атамаў (ад 3—4 да некалькіх дзесяткаў і нават соцень) аднаго ці розных элементаў. Устойлівыя толькі ў саставе
У
Літ.:
Петров Ю.И. Кластеры и малые частицы.
Губин С.П.
В.В.Свірыдаў.
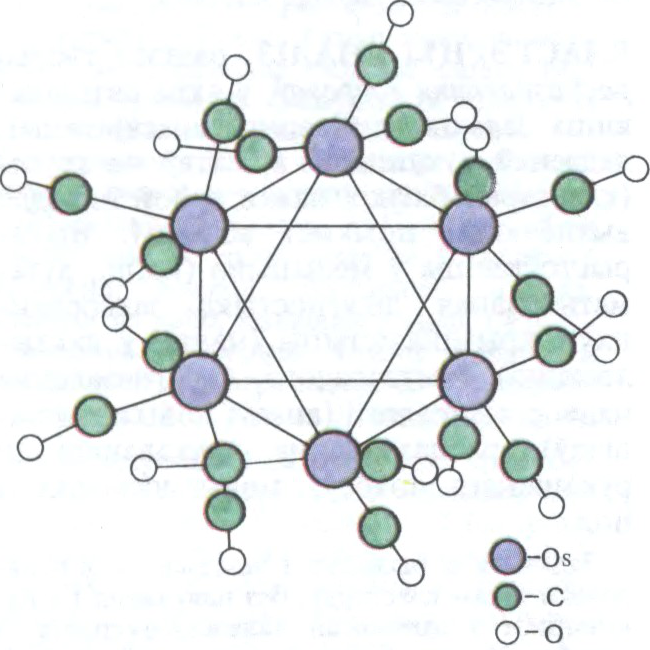
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
КО́БАЛЬТ (
Co, хімічны элемент VIII групы
Бледна-жоўты метал з ружовым ці сіняватым адценнем, tпл 1494
Літ.:
Перельман Ф.М., Зворыкин А.Я. Кобальт и никель.
Большаков К.А.
В.В.Свірыдаў.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
НЕРНСТ ((Nernst) Вальтэр Фрыдрых Герман) (25.6.1864,
нямецкі фізік і фізікахімік, адзін са стваральнікаў фізічнай хіміі.
Тв.:
Літ.:
Гельфер Я.М. История и методология термодинамики и статистической физики. 2 изд.
Льоцци М. История физики:
А.І.Болсун.

Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
ЛІ́ШТВАН (Іван Іванавіч) (
Тв.:
Микро- и макрореология дисперсных систем.
Физико-химические основы технологии торфяного производства.
Физика и
Физические процессы в торфяных залежах.
Массоперенос в природных дисперсных системах.

Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
КАЎЧУ́КІ СІНТЭТЫ́ЧНЫЯ,
эластычныя
Прыняты класіфікацыя і назва К.с. па манамерах, якія выкарыстоўваюць для атрымання каўчукоў (ізапрэнавыя, бутадыенавыя, бутадыен-стырольныя і да т.
Літ.:
Догадкин Б.А., Донцов А.А., Шершнев В.А.
Синтетический каучук. 2 изд
Я.І.Шчарбіна.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
КАЗЛО́Ў (Мікалай Сямёнавіч) (17.5.1907,
Тв.:
5,6-Бензохинолины.
Катализаторы риформинга.
Каталитические свойства соединений редкоземельных металлов.
Ультрастабильные цеолиты.
Літ.:
Н.С.Козлов // Весці

Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
КАПУ́ЦКІ (Фёдар Мікалаевіч) (
Тв.:
Лекарственные препараты на основе производных целлюлозы.
Катионный олигомер пиперилена: синтез, свойства и применение.
Літ.:
Ф.Н.Капуцкий // Вестн. БГУ. Сер. 2.
П.М.Бараноўскі.

Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
ПАХУ́ЧЫЯ РЭ́ЧЫВЫ,
арганічныя злучэнні, якія маюць прыемны пах і выкарыстоўваюцца ў вытв-сці парфумерных і касметычных вырабаў, тавараў
Да 19
Асн. колькасць П.р., што маюць
Літ.:
Братус И.Н.
Я.Г.Міляшкевіч.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)
АКІСЛЕ́ННЕ БІЯЛАГІ́ЧНАЕ,
біяхімічны працэс, сукупнасць акісляльна-аднаўляльных рэакцый. Адбываецца ва ўсіх жывых клетках (пераважна ў мітахондрыях), складае аснову тканкавага дыхання і браджэння.
Вывучэнне працэсаў акіслення ў арганізме пачалося ў 18
На Беларусі розныя аспекты акіслення біялагічнага вывучаюць у ін-тах біяарганічнай хіміі, фотабіялогіі, фізіялогіі, біяхіміі (Гродна)
Літ.:
Кривобокова С.С. Биологическое окисление: Ист. очерк
Ленинджер А. Основы биохимии:
Строев Е.А. Биологическая
Березов Т.Т., Коровкин Б.Ф. Биологическая
М.М.Філімонаў.
Беларуская Энцыклапедыя (1996—2004, правапіс да 2008 г., часткова)